Study design
Design
The CASES trial is a phase 3 international multicenter randomized clinical trial with open label treatment and blinded outcome assessment (PROBE design). The study will have a non-inferiority design. The study will be performed among thrombectomy capable stroke centers in the Netherlands and Belgium.
Population
Inclusion criteria:
The study population will consist of adult patients who meet the following inclusion criteria:
- Acute ischemic stroke due to proximal intracranial occlusion in the anterior circulation (intracranial ICA, M1, proximal M2) on the CT angiography
- Stenosis >50% according to the NASCET criteria16 or initial occlusion of the ipsilateral cervical carotid artery of presumed atherosclerotic origin on baseline CT angiography
- Eligible for EVT according to the guidelines: EVT within 6 hours of onset or EVT between 6-24 hours after onset based on perfusion CT imaging selection (conform current guidelines)
- Baseline National Institute of Health Stroke Scale (NIHSS) score ≥ 2
- Age >18 years
- Written informed consent (deferred consent)
Exclusion criteria:
A potential subject who meets any of the following criteria will be excluded from participation in this study:
- Any intracranial haemorrhage
- Cervical carotid artery stenosis or occlusion with other causes than presumed atherosclerosis (e.g. carotid artery dissection, floating thrombus, carotid web)
- Any exclusion criterion for EVT according to the guidelines
- Pre stroke disability (defined as a modified Rankin Scale score >2)
- Recent gastro-intestinal or urinary tract haemorrhage (<6 weeks)
- Recent severe head trauma (<6 weeks)
- Recent infarction on baseline brain CT in the same vascular territory (< 6 weeks)
- Known allergy to aspirin and/or clopidogrel
- Pregnancy
- Participation in another randomized controlled intervention (EVT) trial
- Alberta Stroke Program Early CT Score (ASPECTS) <6
Randomization
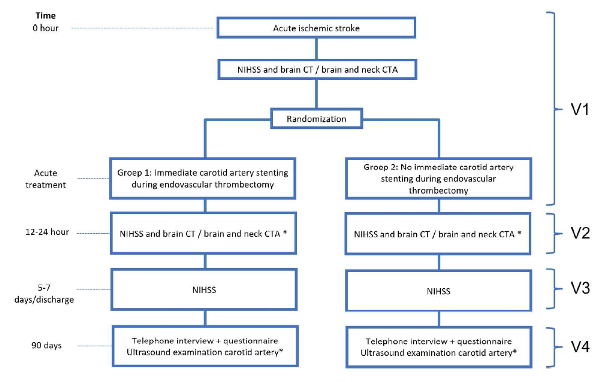
Patients will be randomly allocated to immediate carotid artery stenting during endovascular treatment or the deferred treatment approach: carotid artery endarterectomy, stenting or best medical therapy alone, based on functional recovery after stroke.
Primary outcome
The main study outcome is functional outcome at 90 days post onset as measured on the modified Ranking Scale score.
Sample size
600 patients
Trial duration
36 months
Flowchart of study procedures
Informed consent
This trial will include patients using a deferred written informed consent procedure. This means that, after the treatment, written informed consent is obtained by asking the patient, or a legal representative of the patient.
The justification for the deferred consent is as follows.
Firstly, during acute ischemic stroke, the adagium “time is brain” holds. Reperfusion therapy should be initiated as soon as possible. For every hour delay, the chance of recovery to activities of daily living (ADL) independency decreases with 6%. Secondly, during the acute phase of acute ischemic stroke a patient is often unable to make a well thought out decision on whether to participate in a study, due to neurological deficits like language disturbances, unable to communicate or anosognosia (unaware of his or her medical condition). Obtaining consent from the patient’s proxy will also unacceptably delay the EVT itself.
After treatment, informed consent will be obtained by asking the patient or a legal representative of the patient, preferably within 24 hours after EVT. The patient and/or representative will be provided with a verbal and written explanation of the study by the local investigator. They will be asked for consent to use their data, biomaterials for the purpose of the study and to perform follow up investigations. Participation in the study is voluntary and at any given time, informed consent can be withdrawn.